Esta web utiliza cookies para que podamos ofrecerte la mejor experiencia de usuario posible. La información de las cookies se almacena en tu navegador y realiza funciones tales como reconocerte cuando vuelves a nuestra web o ayudar a nuestro equipo a comprender qué secciones de la web encuentras más interesantes y útiles.

La hemofilia es una coagulopatía (un problema de sangrado) poco frecuente. Las personas con hemofilia no tienen suficiente factor de coagulación en su sangre.
La hemofilia es causada por una alteración en los genes F8 o F9 que producen el factor VIII (FVIII) y el factor IX (FIX) de la coagulación, se trata de una enfermedad producida por la deficiencia de uno de estos factores en el sistema de coagulación.
El sistema de coagulación funciona gracias a 13 factores (Factor I, Factor II, Factor III, Factor IV, Factor V, Factor VI, Factor VII, Factor VIII, Factor IX, Factor X, Factor XI, Factor XII y Factor XIII) coagulantes que trabajan conjuntamente en lo que se llama la “cascada de coagulación”. Si uno de estos factores no funciona bien, la cascada se interrumpe y se forma más lentamente el coágulo que impide el sangrado. Como consecuencia de esta interrupción en la cascada de coagulación, las lesiones o heridas sangran durante más tiempo del debido, pudiéndose producir hemorragias internas y externas.
Es una enfermedad que no se contagia y que afecta a 1 entre 10.000 casos en la población.
Se han descrito los déficits congénitos de diferentes factores de la coagulación factor II, factor V, factor VII, factor X, pero identificamos con Hemofilia el déficit de factor VIII y factor FIX.
Hemofilia A (Factor VIII): Es el tipo de hemofilia más conocida y afecta a 1 entre 10.000 casos en la población.
Hemofilia B (Factor IX): También llamada enfermedad de Christmas, es el tipo menos frecuente y afecta a 1 entre 50.000 casos en la población.
La hemofilia generalmente se hereda, lo que significa que se transmite a través de los genes de los padres. Los genes transmiten mensajes sobre la forma en que se desarrollarán las células del cuerpo a medida que un bebé se convierte en adulto. Determinan el color de cabello y ojos de una persona, por ejemplo.
Los cromosomas procedentes del padre y de la madre contienen la información genética que define cómo somos y si tendremos alguna patología hereditaria. Y dentro de los cromosomas están los llamados cromosomas sexuales que definen el sexo, son los cromosomas X e Y. La herencia genética de la hemofilia se asocia al cromosoma X. En el hombre, al tener sólo un cromosoma X (XY) si éste está dañado, tendrá la enfermedad ya que su otro cromosoma, el Y, no tiene capacidad para producir factor VIII o factor IX.
En la mujer, se poseen dos cromosomas XX. Aunque uno de los dos cromosomas sea portador de una anomalía, se producirá factor VIII o factor IX gracias a la existencia de otro cromosoma X normal. Es decir, en el caso de que una mujer tenga una copia del gen alterado de cualquiera de sus padres, se dice que “porta” el gen de la hemofilia y, por lo tanto, se la llama “portadora”. En otras palabras, tiene una copia normal y una alterada del gen
En este caso nos encontramos con una mujer portadora de la enfermedad. En promedio, las portadoras de hemofilia tendrán alrededor del 50 por ciento de la cantidad normal de factor de coagulación, pero algunas portadoras tienen niveles mucho más bajos de factor de coagulación.
Los hombres no pueden ser meros portadores, si su cromosoma X está dañado, siempre van a manifestar la enfermedad.
En el caso más frecuente de mujeres no portadoras y hombre con hemofilia, los hijos no tendrán la enfermedad y las hijas serán todas portadoras.
En el caso de que una mujer portadora puede transmitir cualquier gen a sus hijos e hijas. Por cada nacimiento, hay un 50 % de posibilidades de que un hijo tenga hemofilia y un 50 % de posibilidades de que una hija sea portadora del gen.
Haga clic aquí para obtener más información sobre personas portadoras y mujeres con hemofilia.
La siguiente figura explica cómo se hereda el gen de la hemofilia en los tres casos más comunes.
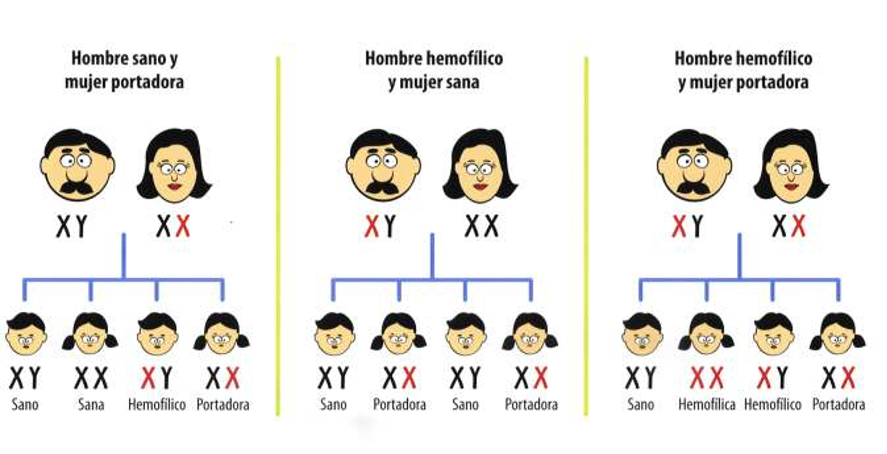
La transmisión de la Hemofilia se dice que es recesiva y no dominante ya que puede que no aparezca en una generación siguiente (salto de generación) por la simple razón de que se den portadoras sanas o varones sanos, y sí aparezca en otra generación posterior
A veces, la hemofilia puede ocurrir cuando no hay antecedentes familiares de la misma. Esto se llama hemofilia esporádica . Alrededor del 30% de las personas con hemofilia no la contrajeron a través de los genes de sus padres. Fue causado por un cambio en los propios genes de la persona.
Hemofilia adquirida
En casos raros, una persona puede desarrollar hemofilia en el futuro. La mayoría de los casos involucran a personas de mediana edad o ancianas, o mujeres jóvenes que han dado a luz recientemente o se encuentran en las últimas etapas del embarazo. Esta condición a menudo se resuelve con el tratamiento adecuado.
El fenotipo característico de la hemofilia es la tendencia a las hemorragias
Hemorragia es la pérdida de sangre que puede ser:
- Interna: Es de decir se producen dentro de las articulaciones y los músculos (rodillas, los codos, y los tobillos, así como los músculos del brazo superior y del antebrazo, el músculo de psoas, del muslo, y de la pantorrilla.). Las hemorragias internas ocurren con mayor frecuencia que las externas, pero éstas no siempre son apreciables.
- Externa: Se produce por un orificio natural del cuerpo (como la nariz, boca, oídos, etc.) o a través de una herida.
Si hay sangrado repetidas veces en una misma articulación, dicha articulación puede dañarse y provocar dolor.
Las hemorragias repetidas pueden causar otros problemas de salud, como artritis. Esto puede provocar dificultad para caminar o para realizar actividades sencillas. Sin embargo, las articulaciones de las manos generalmente no están afectadas en la hemofilia (a diferencia de lo que ocurre en algunos tipos de artritis).
El diagnóstico de la hemofilia se realiza tomando una muestra de sangre y midiendo el grado de actividad del factor. La hemofilia A se diagnostica haciendo pruebas del grado de actividad de coagulación del factor VIII. La hemofilia B se diagnostica midiendo el grado de actividad del factor IX.
Según la cantidad de factor deficitario en el factor VIII o IX se pueden establecer los grados de severidad.
| Nivel | Porcentaje de actividad normal de factor en la sangre | Número de unidades internacionales (UI) por mililitro (ml) de sangre entera |
| Rango normal | 50%-150% | 0.50–1.5 IU |
| Hemofilia leve | 5%-40% | 0.05–0.40 IU |
| Hemofilia moderada | 1%-5% | 0.01–0.05 IU |
| Hemofilia severa | por debajo del 1% | por debajo del 0.01 IU |
Las manifestaciones hemorrágicas están en relación con la gravedad de la hemofilia.
Las personas con hemofilia leve por lo general suelen tener de hemorragias a consecuencia de cirugías o lesiones graves. Podrían nunca llegar a tener un problema de sangrado.
Las personas con hemofilia moderada tienen hemorragias con menos frecuencia, sobre una vez al mes. Pueden sangrar durante mucho tiempo tras una cirugía, una lesión seria, o procedimientos odontológicos. Rara vez, sangran sin que haya un motivo claro.
Las personas con hemofilia severa tienen de hemorragias frecuentes en músculos o articulaciones. Sin tratamiento preventivo, pueden sangrar una o dos veces por semana. La hemorragia es con frecuencia espontánea, lo que quiere decir que ocurre sin causa aparente.
Estas pruebas pueden realizarse en un centro de tratamiento de hemofilia, que podéis encontrar en el siguiente enlace: Centros de Tratamiento de Hemofilia en España
La hemofilia es una enfermedad que requiere de un tratamiento y cuidados específicos para que las personas gocen de un buen estado de salud, a pesar de la coagulopatía.
La cantidad de medicación y la frecuencia con que debe ser administrada, varía de unos a otros tratamientos a otros, también depende del tipo de coagulopatía, el grado de severidad de la misma, del tipo de hemorragia y de su localización.
El propio afectado puede administrarse la medicación previas indicaciones de los profesionales sanitarios, siendo la edad recomendada para iniciarse en esta práctica, los ocho años. La posibilidad de autotratamiento está. regulado por la Resolución del 28 de Abril de 1982 de la Subsecretaría de Sanidad (BOE 02/06/1982, núm.131).
El autotratamiento consiste en que el propio afectado de hemofilia o un familiar administre por vía intravenosa el factor de coagulación deficiente en el propio domicilio y bajo un protocolo higiénico y de pasos de actuación, lo que les proporciona una mayor autonomía frente a los centros sanitarios y una menor dependencia y limitación ante su enfermedad.
Opciones de tratamiento
Los concentrados de factor de coagulación: Constituyen el tratamiento preferido para la hemofilia, se diferencian en dos tipos de acuerdo a su fuente biológica de obtención:
Productos derivados de plasma (Hemoderivados, o plasmáticos): Pueden fabricarse a partir de sangre humana.
Productos recombinantes: Utilizando células genéticamente diseñadas que portan un gen de factor humano.
Los concentrados de factor se elaboran en sofisticadas instalaciones de fabricación. Todos los concentrados de factor de preparación comercial son tratados para eliminar o inactivar virus transportados por la sangre.
El crioprecipitado: Es derivado de la sangre y contiene una moderada concentración de factor VIII (pero no de factor IX). Es eficaz para hemorragias articulares y musculares, pero menos seguro que los concentrados en términos de contaminación viral; además, su almacenamiento y administración son más difíciles. El crioprecipitado puede elaborarse en instalaciones locales de recolección de sangre.
El plasma fresco congelado (PFC): Del que se han eliminado los glóbulos rojos dejando las proteínas sanguíneas y los factores de coagulación es menos eficaz que el crioprecipitado para el tratamiento de la hemofilia A porque el factor VIII está menos concentrado. Es necesario transfundir grandes volúmenes de plasma. Esto puede causar una sobrecarga circulatoria.
Desmopresina (DDAVP): Las personas con hemofilia A leve a veces utilizan, una hormona sintética que estimula la liberación de factor VIII, para tratar sangrados menores.
Anticuerpos terapéuticos: moléculas creadas en un laboratorio para tratar enfermedades, entre ellas la hemofilia, se administran por vía subcutánea.
Actualmente en el mercado están HEMLIBRA que recibió la aprobación de la FDA en el 2017 como profilaxis para las personas con hemofilia A con inhibidores del factor VIII y en el 2018 como profilaxis para las personas sin inhibidores del factor VIII.
Tratamiento a demanda
Es el tratamiento utilizado una vez que se ha producido la herida o hemorragia. En este caso si fuera necesaria la medicación, ésta se administra en la dosis y periodicidad adecuada, prescrita por equipo especialista para detener la hemorragia.
Tratamiento en profilaxis
La profilaxis en hemofilia se basa en la observación clínica de que las personas enfermas con déficit moderados de factor VIII o factor IX no tienen hemorragias frecuentes y principalmente porque no suelen desarrollar patología articular severa.
Mediante la administración de la medicación varias veces por semana (Dependiendo de la coagulopatía y la terapia prescrita), es posible mantener unos niveles plasmáticos mínimos superiores al 1 -3%. Con ello se consigue la protección necesaria para prevenir hemorragias espontaneas o secundarias o traumatismos leves.
En el caso de profilaxis con factor de coagulación, las dosificaciones propuestas se basan en dos protocolos:
El protocolo de Malmö: Aplicaciones de 25-40 UI/kg, administradas tres veces a la semana a quienes tienen hemofilia A, y dos veces a la semana a quienes tienen hemofilia B.
El protocolo de Utrecht: Aplicaciones de 15-30 UI/kg, administradas tres veces a la semana a quienes tienen hemofilia A, y dos veces a la semana a quienes tienen hemofilia B
Sin embargo, no se ha establecido un protocolo definitivo. En la medida de lo posible, éste debería adaptarse a las necesidades y circunstancias individuales de cada persona, también a los productos disponibles, ya que en la actualidad existen concentrados de factor de vida media extendida y de larga duración y productos subcutáneos que nos permiten ajustar la profilaxis
Hay varios tipos de profilaxis. La profilaxis continua (primaria, secundaria y terciaria) se administra de manera periódica durante varios meses e incluso años. La profilaxis intermitente o periódica se administra durante periodos más breves, generalmente por unas cuantas semanas o meses.
| Tipos de Profilaxis | Definición |
Profilaxis primaria | Tratamiento periódico continúo iniciado antes de la segunda hemorragia en una articulación mayor y antes de cumplir los 3 años. |
| Profilaxis secundaria | Tratamiento periódico continúo iniciado después de dos o más hemorragias en articulaciones mayores, pero antes de la aparición de la enfermedad articular. |
| Profilaxis terciaria | Tratamiento periódico continuo iniciado después de la aparición de la enfermedad articular para evitar la progresión del daño. |
| Profilaxis intermitente (“periódica”) | Tratamiento administrado para evitar hemorragias durante periodos cortos; por ejemplo, antes y después de una cirugía. |
¿Qué es un PORT-A-CATH?
Es un reservorio subcutáneo implantado bajo la piel mediante una pequeña intervención quirúrgica, está conectado a una vena profunda de la zona subclavia (debajo de la clavícula). Está indicado sobre todo en niños/as con Hemofilia con un difícil acceso para la administración del factor en las venas, así como en edades tempranas para facilitar un tratamiento en profilaxis. El implante de un PORT-A-CATH debe discutirse ampliamente, dado que existe riesgo de hemorragia e infección.
¿Qué es una tunelización?
Consiste en canalizar siempre la misma vena utilizando el mismo punto de inyección. Se crea una especie de túnel o “fístula” artificial que con el tiempo facilita el acceso venoso. Está recomendado en niños ya que la tunelización minimiza el dolor de la venopunción, asegurando el cumplimiento del tratamiento y favorece los autocuidados.
La OMS (Organización Mundial de la Salud) define la adherencia al tratamiento como el cumplimiento del mismo, es decir toma la medicación de acuerdo con la dosificación y el programa prescrito. Pero además se considera la persistencia, que es tomar la medicación a lo largo del tiempo del tratamiento indicado. Según la Organización Mundial de la Salud, los porcentajes de falta de adherencia de cualquier tratamiento farmacológico varían entre 15% y 93%, con un porcentaje promedio calculado en 50%.
La adherencia al tratamiento es vital en la profilaxis, ya que el éxito depende de mantener los niveles de factor continuamente por arriba del nivel objetivo. Especialmente desde la infancia.
Las consecuencias de la falta de adherencia se pueden resumir en un empeoramiento de la calidad de vida del paciente, una falta de control de la enfermedad, una mayor probabilidad de recaídas y agravamientos, la aparición de efectos secundarios o intoxicaciones e incluso aumentar la morbilidad. Pero es que, además, puede suponer una falta de racionalización en el gasto farmacéutico y sanitario y desde un punto de vista médico puede suceder que los tratamientos lleguen a ser ineficaces por la aparición de resistencias o favorecer una mayor agresividad de una determinada enfermedad
Dicho esto, pacientes, industria farmacéutica y profesionales médicos deben colaborar a fin de asegurar una adherencia al tratamiento.